

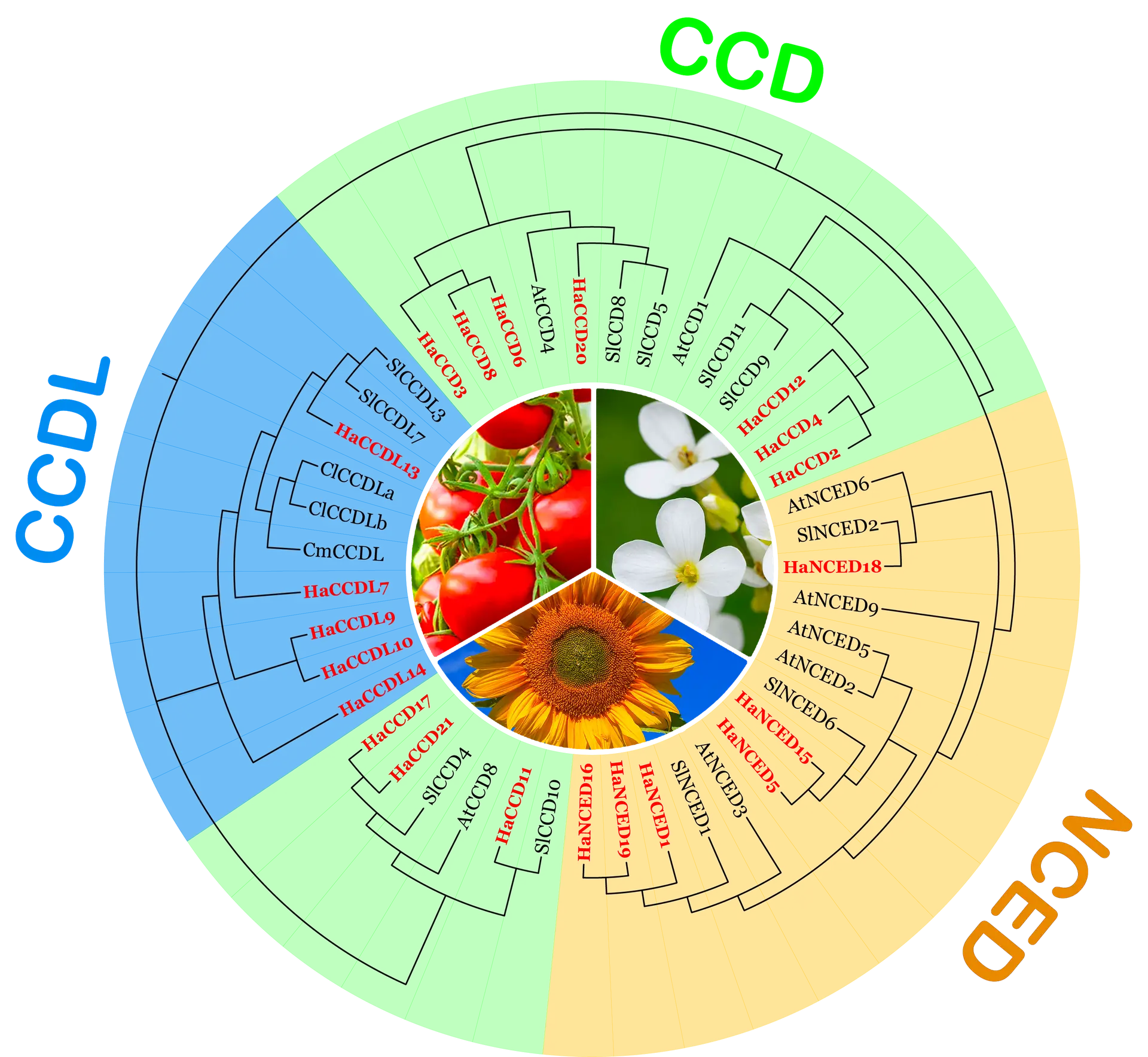
Introduction to Phylogenetic Analysis in Gene Family Analysis
Phylogenetic analysis is a critical aspect of modern genetics that focuses on the classification and evolution of gene family members. A gene family consists of groups of related genes that share a common ancestral gene, exhibiting similar sequences and functions. Understanding gene families is fundamental in deciphering the complexity of genetic interactions and evolutionary history. Genes within a family may evolve through processes such as duplication, divergence, and horizontal gene transfer, resulting in functional diversification. Such diversity holds significant implications for evolutionary biology, comparative genomics, and biotechnology.
The concept of phylogeny refers to the evolutionary history and relationships among various biological species or entities. In phylogenetic analysis, researchers construct phylogenetic trees, which visually represent these evolutionary connections. These trees are formed by analyzing genetic data, such as DNA sequences, and using computational methods to infer the relationships among gene family members. Tree-building methods can vary; common approaches include neighbor-joining, maximum likelihood, and Bayesian inference. Each method employs distinct algorithms to estimate the phylogenetic distances that reflect evolutionary changes over time.
Understanding the phylogenetic relationships among gene family members is crucial for several reasons. It allows scientists to trace the evolutionary pathways that have led to the current diversity of genes. Furthermore, insights gleaned from phylogenetic analysis can help in predicting the function of uncharacterized genes, revealing how certain traits and behaviors may have evolved in different organisms. By elucidating these relationships, phylogenetic analysis not only enhances our grasp of genetic variation but also lays the groundwork for advanced studies in evolutionary theory and applications in areas like medicine and agriculture.
Classification of Gene Family Members into Groups and Subgroups
The classification of gene family members into distinct groups and subgroups is a fundamental aspect of phylogenetic analysis, facilitating our understanding of evolutionary relationships among various organisms. This classification is largely undertaken through several methodologies that encompass sequence similarity, functional annotation, and structural characteristics. Each of these strategies plays a pivotal role in establishing a comprehensive framework for categorizing genes that share common evolutionary origins.
Sequence similarity serves as a primary criterion for classification. This method involves comparing nucleotide or protein sequences across different gene family members to identify conserved regions, which can indicate evolutionary relatedness. The vastness of databases such as GenBank allows researchers to utilize tools like BLAST (Basic Local Alignment Search Tool), which efficiently provides insights into related sequences and helps delineate groups based on similarity thresholds.
In addition, functional annotation is integral to classifying gene families, as it associates specific functions with groups of genes. This process may include the identification of metabolic pathways or biological processes wherein these genes are involved. Annotated databases, like Gene Ontology (GO), provide a structured vocabulary that enhances our understanding of gene functions, leading to more precise classification based on physiological roles.
Furthermore, the evaluation of structural characteristics, such as domain architecture and protein conformation, contributes to a deeper understanding of gene family members. Computational tools like Pfam and SMART allow researchers to categorize genes based on identified protein domains, which are indicative of certain functionalities.
Accurate classification of gene family members is crucial not only for genomic studies but also for evolutionary research, as it can shed light on gene duplications, losses, and adaptations over time. Utilizing these various classification systems ensures a holistic approach to phylogenetic analysis, ultimately enhancing our insights into the complexities of genetic evolution.

https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-024-10351-w
Exploring Evolutionary Relationships Among Gene Family Members
The study of evolutionary relationships among gene family members is essential for understanding the complexities of biological systems. Phylogenetic trees serve as a crucial tool in depicting these relationships, illustrating the divergence and convergence of gene traits over time. By constructing these trees, researchers can visually represent how different gene family members are related, shedding light on their evolutionary history and functional similarities. This process often reveals critical insights into the conservation of genetic sequences across various species, highlighting instances where certain traits have been preserved due to evolutionary advantages.
Divergent evolution occurs when two or more gene family members evolve from a common ancestor and develop distinct functions or characteristics over time. This phenomenon is often observed in gene families that undergo significant mutations leading to varied adaptations in their respective environments. Conversely, convergent evolution refers to the process by which unrelated or distantly related organisms independently evolve similar traits—this can be particularly relevant in gene families that have adapted to analogous ecological niches or survival strategies. Such evolutionary dynamics emphasize the importance of both genetic divergence and convergence in the overall evolution of gene families.
Another noteworthy aspect of evolutionary relationships is horizontal gene transfer (HGT), wherein genetic material is exchanged between different species, making it a vital factor in the evolution of gene families. This transfer can lead to the acquisition of new functions and capabilities, significantly altering the evolutionary path of the recipient species. For instance, HGT is commonly observed in prokaryotes, where it can confer antibiotic resistance, profoundly influencing microbial evolution.
Real-world case studies further illustrate these concepts. For example, the analysis of the globin gene family across various vertebrate species reveals both divergent and convergent evolutionary patterns that have allowed these vital proteins to fulfill essential roles in oxygen transport. Such studies underscore the value of phylogenetic analysis in understanding the complexities of gene family evolution and the shared heritage of life.
Identification of Orthologues in Gene Family Members
Orthologues are genes in different species that evolved from a common ancestor through speciation. They typically retain the same function over evolutionary time, making the identification of orthologues essential for understanding gene function preservation and the evolutionary trajectories of gene families. This process is particularly relevant in evolutionary biology, where orthologue identification fosters comparative studies, aiding in the elucidation of molecular mechanisms shared across taxa.
Several approaches are employed to identify orthologues among gene family members, with each method offering unique insights and varying levels of resolution. One popular method is sequence alignment, which compares nucleotide or protein sequences across different species. Tools like BLAST (Basic Local Alignment Search Tool) facilitate this comparison, revealing similarities that may indicate orthologous relationships. The degree of sequence conservation can be correlated with evolutionary distance, allowing researchers to assess functional preservation relative to genetic divergence.
Comparative genomics builds on the principles of sequence alignment by examining whole genomes. This approach allows for a broader perspective, identifying conserved synteny and gene arrangement that can reinforce the inference of orthology. By analyzing genetic context within extended genomic sequences, researchers can better distinguish orthologues from paralogues (genes that have diverged within the same species).
Phylogenetic methods represent another approach to orthologue identification, where gene trees are constructed based on sequence data to infer evolutionary relationships. By representing genes from multiple species in a phylogenetic framework, scientists can determine which genes are more likely to be orthologues based on their branching patterns.
Despite the advances in techniques for identifying orthologues, challenges remain. Issues such as gene duplication, loss, and incomplete lineage sorting can complicate clarity in orthologous relationships. Thus, employing multiple methods and integrating various datasets is crucial for robust orthologue identification and ensuring reliable conclusions in evolutionary studies.

Motif And Domain Analysis Of Protein In Genome Ide Analysis
[…] Step 2 Phylogenetic analysis roles in the classification and evolution of Gene Family Members […]